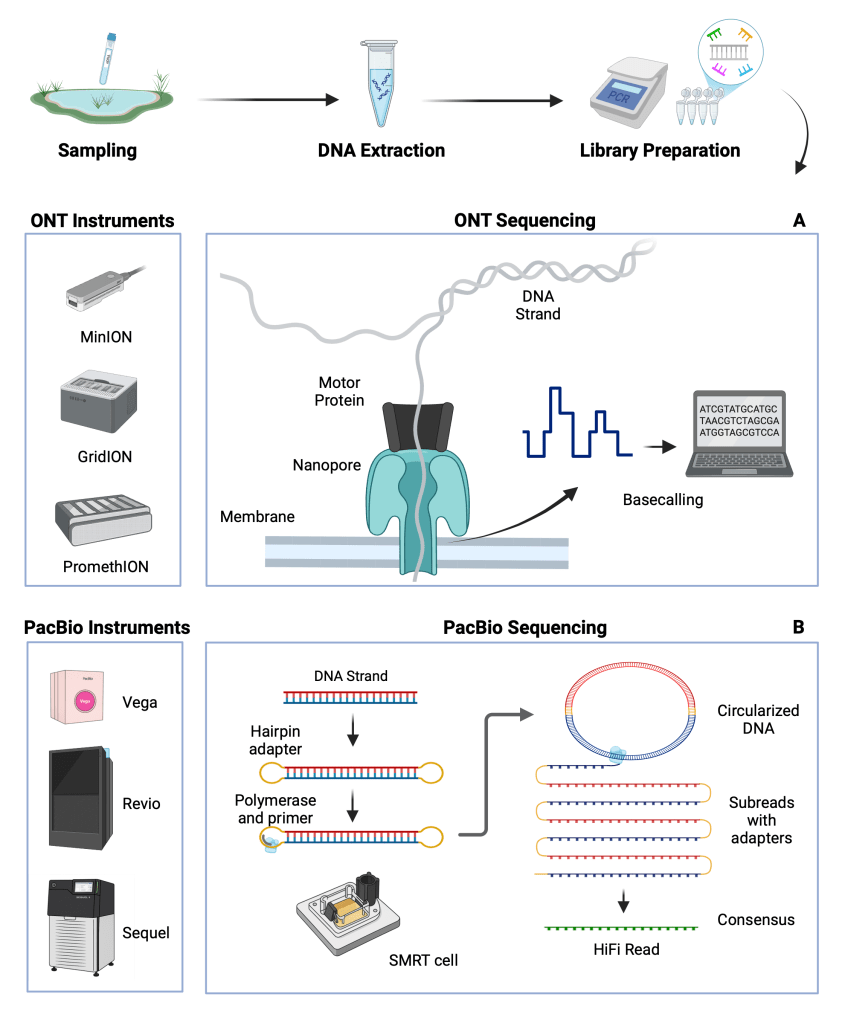
DNA-based biodiversity monitoring
The use of DNA-based methods for species detection has revolutionised biodiversity monitoring offering increased speed and accuracy of detection, while reducing disturbance caused by specimen sampling. DNA extracted from environmental samples (eDNA) can be used for monitoring of different types of ecosystems, while co-extraction of multiple specimens (community samples) can also be used to reduce processing time and increase throughput.
Long-reads for biodiversity monitoring
The increasing impact of anthropogenic activities is causing rapid loss of biodiversity and presses the need for continued development of improved methods for biodiversity monitoring. DNA-based monitoring applications have greatly advanced the way we see and understand biodiversity.
Recently, the development of long-read sequencing technologies has created novel applications and introduced a new and exciting field of biodiversity analysis.
Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) long-read sequencing platforms and their advanced capabilities can provide novel solutions to understanding and measuring biodiversity and advance the existing DNA-based monitoring tools.
Freshwater ecosystem monitoring using eDNA time-series
Freshwater ecosystems are increasingly under threat due to anthropogenic activities, and eDNA based applications have been particularly developed to advance lake and river monitoring to meet national and international directives.
Using different sample types including water samples and invertebrate communities we can monitor diversity in aquatic ecosystems more efficiently than with traditional morphological approaches.
Aquatic macroinvertebrate taxa are used as bioindicators including non-bitting midges (Chironomidae), caddisflies (Trichoptera), stoneflies (Plecoptera), and mayflies (Ephemeroptera).

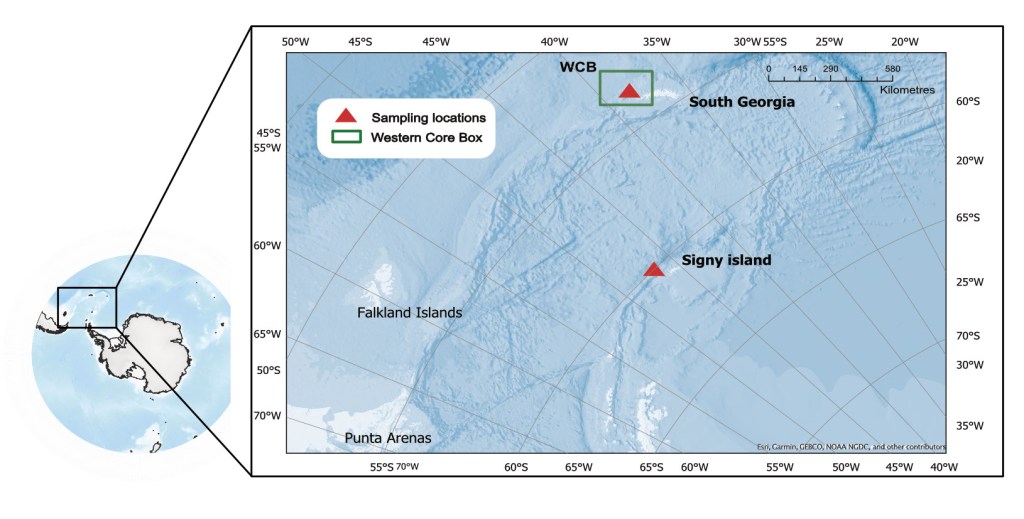
The Southern Ocean eDNA project
Antarctica is one of the fastest warming regions on Earth. The effects of climate change could impact thermo-sensitive populations of fish, such as the extremely stenothermic notothenioids. Using eDNA we can monitor species diversity and potentially identify population structure which will be very useful for informing management decisions.
In the Southern Ocean eDNA project (SO eDNA), we use water extracted eDNA samples coupled with whole genome data to investigate population structure of fish populations.

Mito-metagenomics improves biomass estimations of invertebrate communities
PCR-based analysis of community samples, such as metabarcoding, can introduce bias in biomass estimations. Using PCR-free shotgun sequencing we can improve biomass estimations, which is necessary for accurate ecosystem assessment.
In Bista et al. 2018, we used macroinvertebrate community samples of known biomass to compare the efficiency of each method with regards to accuracy of biomass estimation. Mito-metagenomic sequencing outperformed metabarcoding in biomass estimation. Combining results from multiple amplicons improved the accuracy of the metabarcoding approach.

Comparative genomics
In comparative genomics we use whole genome sequences of different species to perform comparisons of their genomic features. With the aid of bioinformatics tools we can study differences and similarities between species and illuminate their evolutionary history and mechanisms of adaptation to different environments . Some of the studied features are conserved genes and genomic regions, structural variants, novel genes etc. The recent advances in whole genome sequencing have provided a boost in comparative genomics studies. We are using comparative genomics to study the evolutionary history of different taxa, especially fish and other aquatic animals.
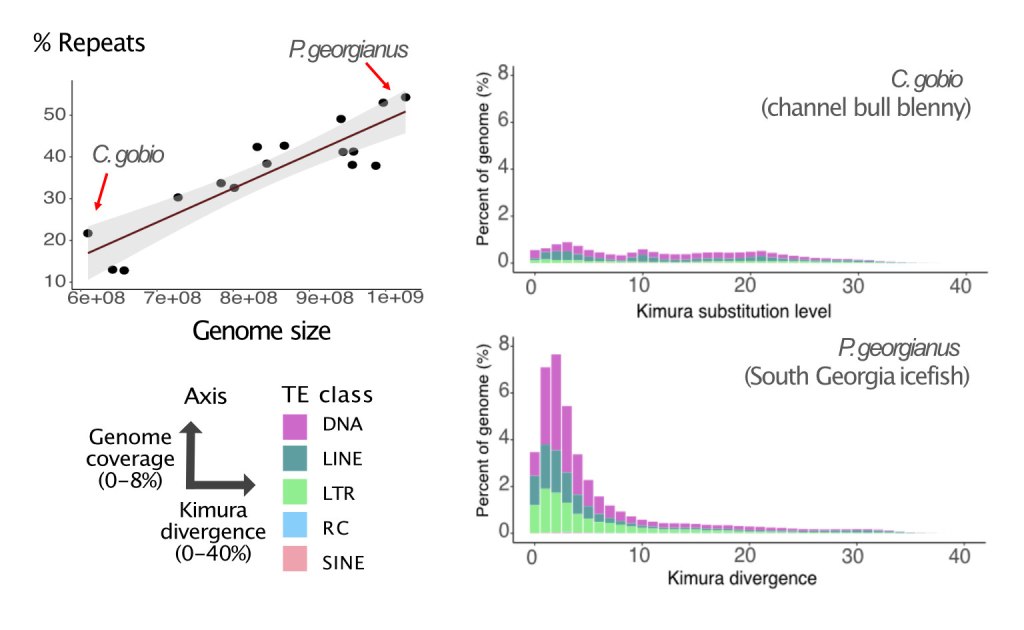
Antarctic notothenioid fish genomes project
Notothenioids are an important marine fish adaptive radiation native to the waters of the Southern Ocean in Antarctic and sub Antarctic regions. They are adapted to life in extreme cold using adaptations such as antifreeze glycoproteins. To understand the mechanisms driving genomic adaptation to cold in this charismatic fish group, we generated and analysed 24 new genome assemblies, including five reference genome assemblies. Using this data we were able to reconstruct the structure of two highly complex gene families, the antifreeze and the haemoglobin genes, generate a new dated phylogeny, and link a genome size increase (~65%) to massive expansion of transposable elements.

Transposable elements in genomic evolution
Transposable elements (TEs) are powerful facilitators of genomic evolution, and can be responsible for loss or gain of function. The recent improvements in generation and availability of reference genomes with properly assembled repeat regions has enabled deep characterisation of transposon diversity. We are interested in investigating the effects of transposable elements in species adaptation, and have been working on TE analysis in diversity of species, such as fish (Bista et al. (2023), Kwon et al. 2022), fungi (Dellaire et al. 2021), and currently invertebrate species.
Evolution and diversity of subterranean species
The unique nature of subterranean environments has facilitated the evolution of highly specialised organisms adapted to survival in extreme conditions. Species adapted to cave environments exhibit characteristic troglomorphies which refer to a set of convergent adaptive phenotypes associated with cave environments, such as loss of pigmentation and reduction or loss of vision. Using subterranean fauna as target and the unique characteristics of caves as eco-evolutionary laboratories we explore the mechanisms of adaptation to challenging environmental conditions.
Due to the nature of subterranean habitats, species that inhabit these ecosystems are often rare, hard to collect and difficult to identify. To improve our knowledge of subterranean diversity we are working on improving DNA based monitoring of threatened subterranean communities.

Reference genome sequencing
Vertebrate Genomes Project (VGP)
Generation of high-quality reference genomes has revolutionised studies on species ecology and evolution. I have been working as contributor of genomics initiatives such as the Vertebrate Genomes Project to advance methods for reference genome generation across the tree of life (Rhie et al. 2021).
In order to enable high-throughput generation of high quality reference genome assemblies large efforts are required in the development of methodologies for long-read sequencing. At the Sanger Institute in collaboration with the VGP, I have been working towards testing and improving methods for tissue preservation, extraction, and sequencing for optimised reference genome sequencing (Dahn et al. 2022).

European Reference Genome Atlas (ERGA)
The European Reference Genome Atlas (ERGA) is a pan-European initiative aiming to advance collaboration between European researchers, and promote generation of reference genomes for study of non-model eukaryote species (Formenti et al. 2022).
Working in collaboration with ERGA, the Sanger Institute, and University of Ioannina we have generated the first genome assembly as part of the ERGA pilot project. This work aims at generation and analysis of a reference assembly for Aristotle’s catfish, which endemic to NW Greece.

Unknown Germany – An integrative biodiversity discovery program

Selected ongoing collaborations
Prof. Melody Clark, British Antarctic Survey: extremophile sequencing, fish genomics, Antarctic biodiversity
Prof. Richard Durbin, and Prof. Eric Miska University of Cambridge: computational genomics, comparative genomics. Reference genome sequencing of fish diversity. Contribution of transposable elements in genomic adaptation.
European Reference Genome Atlas: Reference genome of Aristotle catfish and other Greek vertebrate species as part of the ERGA Pilot project



Contact
Dr. Iliana Bista, Senckenberg – LOEWE TBG
Senckenberganlage 25, 60325, Frankfurt, Germany
iliana.bista@senckenberg.de
